1、欧盟的三类提前审评药品流程
欧盟没有单独的紧急使用授权制度,以一般药品授权的评审流程(见图1-1)为基础,欧盟理事会(EC)与欧洲药品管理局(EMA)根据《欧盟理事会条例第726条/2004》[Regulation(EC)No726 /2004]设立了三种评审程序:加速审评,附条件上市许可和特殊审评(见图1-2)。后于Reg.507 /2006 /EC中规定,附条件上市许可从属于加速审评。
1.1 加速审评
Reg.726 /2004 /EC第14(9)条加速审评(Acceleratedassessment)规定,从公共卫生特别是从治疗创新角度,申请人可申请加速审评,审评时限由最短210天缩减至最短150天。Reg.726 /2004/EC条例规定,“为满足特定患者的合理期望,并考虑到科学和治疗方法的日益迅速发展,应建立加速审评程序,保留具有重大治疗利益药品以及依据年度审查条件获得临时许可(TemporaryAuthorization)的审评程序”。
其中重大公共卫生利益药品指的是由申请人证明并由欧盟人用药品委员会(CHMP)根据具体情况评估药品是否具有重大公共卫生利益,评估标准包括:(1)存在未满足的医疗需求和可用的预防、诊断或治疗方法。(2)药品预期满足现有医疗需求的程度。(3)从公共卫生角度证明药品重大效益的证据强度。
1.2 附条件上市许可和其下的疫苗紧急授权程序
附件性上市许可(Conditionalmarketing authorisation,CMA)根据Reg.726/2004/EC规定,在与申请人协商后,可附加某些特定义务予以批准上市许可。该许可初始有效期为1年,由EMA每年审查允许后续转为普通许可。附条件上市许可适用情况之一是紧急情况下使用的药品,应提供理由说明药品拟在紧急情况下使用,以应对世界卫生组织(WHO)决议或决定或欧盟(EC)正式确认的公共卫生威胁,为响应严重境内或跨境健康威胁采取的措施。
附件性上市许可程序中还包括疫苗紧急授权程序。EMA为快速审批流感大流行期间研发的新疫苗设立了一条途径,可将审评时限由缩减为70天。这一程序允许疫苗上市时数据仍不完整,在申请人承诺提供后续研究结果等相关数据和信息的条件下,批准疫苗上市。疫苗紧急授权程序快速审评采用滚动式审评机制(RollingReview),即分阶段递交生物制品许可申请或新药申请申报材料,无须待所有资料全部完成后再提交审评。
1.3 特殊审评下的模拟疫苗程序(病毒株变更加快审评程序)
特殊审评(Exceptionalcircumstances)是一种针对特定类别药物的审评程序,这些药物通常由于治疗条件的罕见、无法完全收集的信息或不符合伦理要求,因而无法提供出正常使用条件下的疗效或安全性数据。与附件性上市许可不同,特殊审评程序下申请人无需在授权后再向EMA提供全面的药品数据,仅需要安全性和有效性相关的数据。特殊审评下的授权也一般不会转变为标准流程的营销授权。
模拟疫苗是基于备灾目的,是针对当前尚没有大范围流行的流感病毒所生产的疫苗,但其成分和人体使用安全性与最终临床使用的疫苗相符,除流感病毒株不同以外。模拟疫苗程序在申请时不要求提供完整的药学数据,如果风险效益评价结果是正向的,那么可以走特殊审评程序,暂时获得模拟疫苗资格。一旦流感爆发,生产企业可以将模拟疫苗的流感病毒株换成导致流感大流行性的流感病毒株,并且随之开始检测疫苗的安全性和有效性。为了加快审评,生产企业只需要提交与变更的病毒株信息数据相关的变更申请,EMA采用滚动式审评机制审评。EMA科学委员会和CHMP将在5天内向EC提出建议,以获得上市许可最终授权。CHMP给出肯定意见后10~20天,经EC授权,疫苗可立即在临床中使用。授权后仍要对模拟疫苗的安全性和有效性,尤其是对儿童和孕妇的影响进行密切监测,确保疫苗与预期具有相同的安全性和有效性。

图1-1EMA药物评审流程
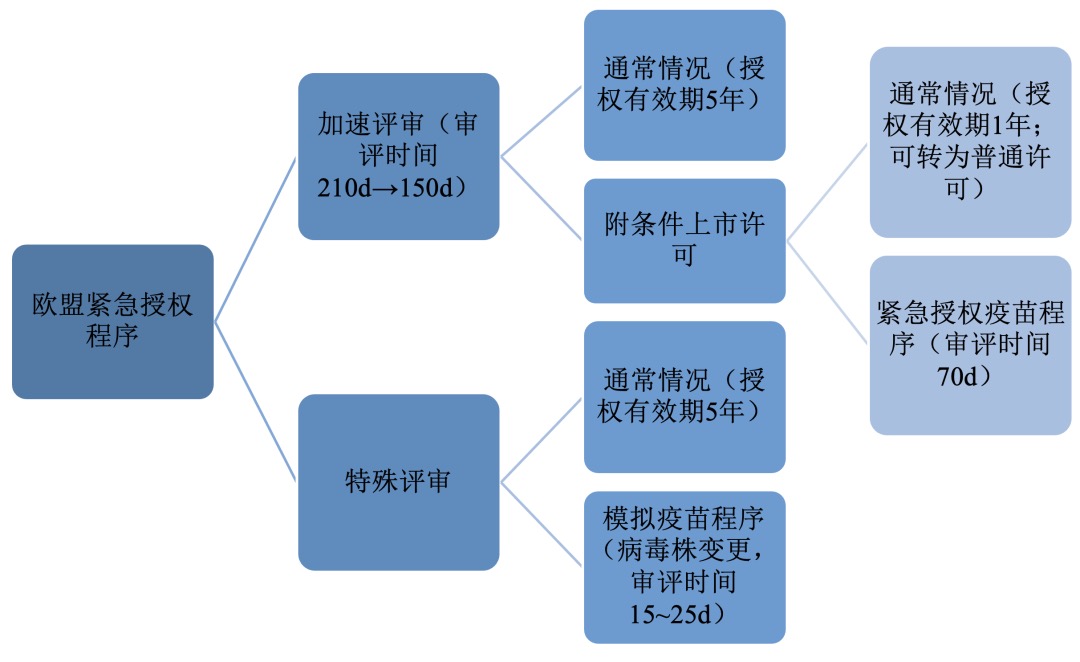
图1-2欧盟紧急授权程序
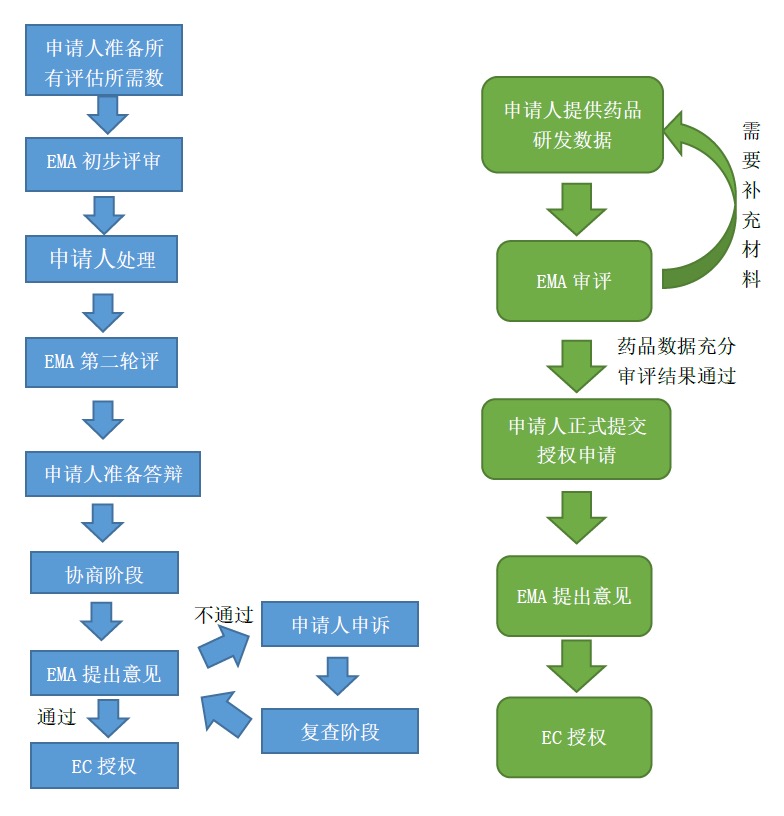
图1-3普通审评流程和滚动审评流程
2、欧盟药品审评案例
2.1 新冠疫苗通过疫苗紧急授权程序申请上市
2020年COVID-19全球大流行期间,辉瑞/BioNTech的BNT-162b2疫苗(Comirnaty疫苗)分别向英国和欧盟提交了紧急使用的授权申请。
英国药品保健品监管机构(MHRA)(是英国脱欧前EMA的主要组成部分之一,成为独立审查机构后审查流程相同)和EMA都从10月6日起进行对疫苗数据的滚动式评审。12月2日,英国依照英国《2012年人类医学法规》第174条通过特殊立法程序临时批准使用辉瑞/BioNTech的BNT-162b2疫苗,成为最早批准新冠疫苗使用的西方国家。欧盟方面的发言人批评英国的措施“仓促而草率”,他们选择了更保守的审评进度,于12月21日完成了对该疫苗的紧急授权疫苗程序予以上市批准。
※注:英国《2012年人类医学法规》第174条规定了四类情况下政府当局可临时批准特殊药品的销售或供应许可。包括了病原体、毒素、化学品和核辐射这四类对人体有健康危害的可疑或已确认的范围性传播情况。
2.2 Zabdeno和Mvabea——一组通过特殊审评获得授权的埃博拉病疫苗
Zabdeno和Mvabea是一组主要针对埃博拉病毒的疫苗,分两剂注射。这两类疫苗的生产背景是于2020年6月爆发于非洲的埃博拉出血热疫情,欧盟向非洲国家提供医疗援助。Zabdeno和Mvabea分别是由腺病毒和改良安卡拉痘疫苗病毒修改而来的含埃博拉病毒蛋白的两种疫苗,通过模拟疫苗程序进行评审。虽然尚未确定针对该病毒的保护水平和持续时间,但欧洲药品管理局认为,疫苗的益处对于帮助控制疫情和预防死亡可能非常重要,且在安全性方面,其大多数副作用的严重程度为轻度至中度,持续时间短。因此,EMA认为Zabdeno和Mvabea疫苗的收益大于其风险,故于当年7月1日起授权其在欧盟使用。
2.3 Onpattro——通过加速审评的孤儿药
Onpattro是首个获得美国FDA和欧盟批准的siRNA药物,用于治疗遗传性转甲状腺素蛋白(hATTR)淀粉样变性引起的神经损伤,于2011年4月15日被指定为孤儿药。EMA认为其符合加速审评的条件(即前文所述的重大公共卫生利益),对Onpattro进行了150天的加速审评后,于2018年8月27日授予其欧盟地区的营销许可。
除了Onpattro外,许多孤儿药也通过加速审评的方式获得授权;但也有由于临床试验过程中的信息不足,故通过特殊评审路径获得授权的孤儿药,例如Vyndaqel,一种治疗家族性淀粉样蛋白多发性神经病的药物。
2.4目前EMA对新冠肺炎相关药品的批准情况
EMA对于与新冠肺炎有关的药品审评流程采取了高透明度的特殊措施,对新药审评过程以高频次的审评信息和数据公布,但不缩短总体审评时间。
2020年中EMA批准了97种药物的营销许可,其中加速审评6种,附条件上市13种,特殊审评5种,和新冠肺炎相关的只有之前提到过的Comirnaty疫苗和一种治疗用药Veklury。截至目前,在欧盟获得授权的新冠疫苗共4种(分别为辉瑞、阿斯利康、莫德纳和强生推出的疫苗);欧盟地区今年新获得营销授权的针对新冠肺炎的新药只有2种(Regkirona与Ronapreve),正在滚动审评中的药物1种(Evusheld),已申请还未开始的审评的药物有5种。
3、小结
EMA通过设立多种审评流程,对药品审评的紧急程度进行了分流。孤儿药、创新药和其他一些针对各类疑难杂症用药,如艾滋病、恶性肿瘤等现代医学的棘手难题,都常常被允许以加速审评甚至特殊审评的路径来提前获得授权,尽快进入医疗市场,让患者得以尽早从新药中获益;而当面对各类公共卫生事件时,EMA则可以通过模拟疫苗程序和紧急疫苗程序来对针对性的疫苗进行授权以尽快面对疫情。
这几类可以加快审评速度的流程既是欧盟对于各类创新药、罕用药的鼓励措施,也保证了欧盟在面对重大公共卫生事件时,审评制度具有一定的紧急应对能力,并保障药品的安全性和有效性。这对于我国药品管理制度的进一步完善有一定的参考价值。但欧盟作为一个跨国家的地区组织,这一套审评程序存在着执行过程僵硬的情况,无法像作为前欧盟成员国的英国那样能通过立法的强硬手段或是美国FDA使用一套成熟的紧急使用授权流程来加快紧急审评这样的方法来尽快推广疫苗以应对危机。欧盟的药品紧急授权过程在全球疫情的背景下显得相对保守,对于COVID-19相关的药品审评过程EMA选择采取了更加透明、频繁的信息公布而非采取压缩时间的方式。


