文丨滴水司南
为指导和规范基因治疗产品的临床试验,2021年12月3日,国家药品监督管理局药品审评中心官网发布《基因治疗产品长期随访临床研究技术指导原则》(成文时间2021年12月1日),自发布之日起实施,本指导原则针对基因治疗长期随访临床研究的观察方法和研究设计进行讨论,着重阐述了基因治疗长期随访临床研究的观察目的、考虑要素、设计实施以及不同基因治疗产品的特殊考虑等相关要求,在为该类产品开展长期随访临床研究提供技术指导,确保及时收集迟发性不良反应的信号,识别并降低这类风险,同时获取这类产品长期安全性和有效性的信息。
为指导和规范基因治疗产品的临床试验,2021年12月3日,国家药品监督管理局药品审评中心官网发布《基因治疗产品长期随访临床研究技术指导原则》(成文时间2021年12月1日),自发布之日起实施,本指导原则针对基因治疗长期随访临床研究的观察方法和研究设计进行讨论,着重阐述了基因治疗长期随访临床研究的观察目的、考虑要素、设计实施以及不同基因治疗产品的特殊考虑等相关要求,在为该类产品开展长期随访临床研究提供技术指导,确保及时收集迟发性不良反应的信号,识别并降低这类风险,同时获取这类产品长期安全性和有效性的信息。

本文为大家梳理了基因治疗产品长期随访临床研究内容并结合自己的解读与大家分享。
PART 01.国内外基因治疗产品长期随访政策简介
尽管基因疗法在治愈疾病方面具有极大潜力,但它也会对人体带来长期或永久性影响,接受基因治疗的患者出现迟发性不良反应的风险可能会增加,如基因治疗产品的活细胞的生物学特性的变化在体内长期存在,可能增加不可预测的风险。为了评估和降低这类风险,并了解治疗效果随时间延长的变化,有必要对参加基因治疗临床试验的受试者开展长期随访。
目前,美国FDA和欧盟EMA均已发布相关技术指导原则,经查询欧盟EMA于2008年发布的《Guideline On Safety And Efficacy Follow-Up-Risk Management Of Advanced Therapy Medicinal Products》和2009年发布《Guideline On Follow-Up Of Patients Administered With Gene Therapy Medicinal Products》指南,为申办方提供了基于基因治疗的风险概况,考量长期随访的指导性建议。
美国FDA于2020年1月发布的《Long Term Follow-Up After Administration Of Human Gene Therapy Products》,对如何设计长期随访研究提出了建议,并对长期随访观测要素、持续时间、数据收集和报告要求等问题提供了指导思路。
考虑到国内尚无相关指导原则对基因治疗产品长期随访临床试验设计进行规范指导,2019年4月,国家药监局启动了中国药品监管科学行动计划,药审中心负责实施的“细胞和基因治疗产品技术评价与监管体系研究”纳入首批研究项目,其中,《基因治疗产品长期随访临床研究技术指导原则》是基因治疗类药物技术评价体系的重要内容,有助于引导基因治疗类药物临床试验的规范开展,CDE在充分调研国内外同品种研发情况以及相关临床试验技术要求基础上,自2021年1月启动,2021年6月发布征求意见稿(会稿截止时间2021年7月4日),2021年12月3日,CDE正式发布《基因治疗产品长期随访临床研究技术指导原则》,自发布之日起实施。
PART 02.如何考虑制定符合基因治疗产品的长期随访策略?
基因治疗产品长期随访的主要目的是收集受试者的迟发性不良反应,了解基因治疗产品在体内的存续情况,从而识别并降低接受基因治疗产品的患者的长期风险。
1、长期随访的持续时间多长合适?
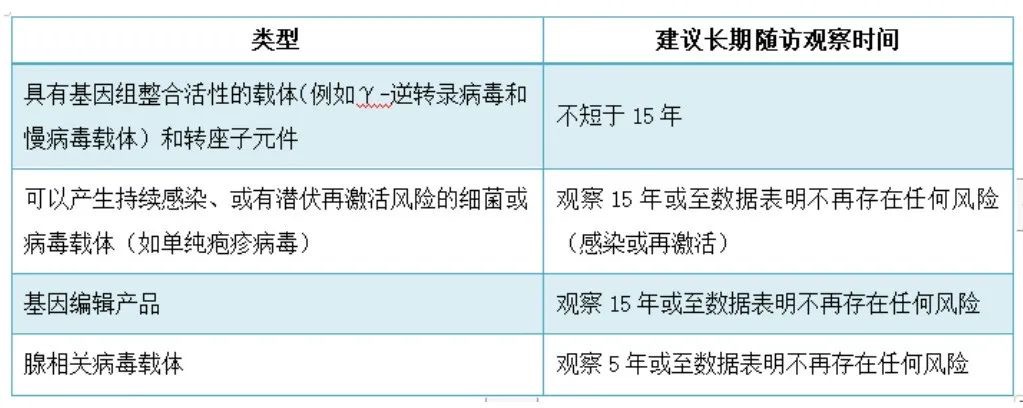
基因治疗作为一种新兴的疗法,其长期安全性仍是未知的,长期随访的持续时间应确保足以观察到受试者因产品特性、暴露情况(生物分布和给药途径)等导致的风险,应不短于迟发性不良反应的预期发生时间。一般而言,针对不同类型的基因治疗产品长期随访观察时间建议如下表:

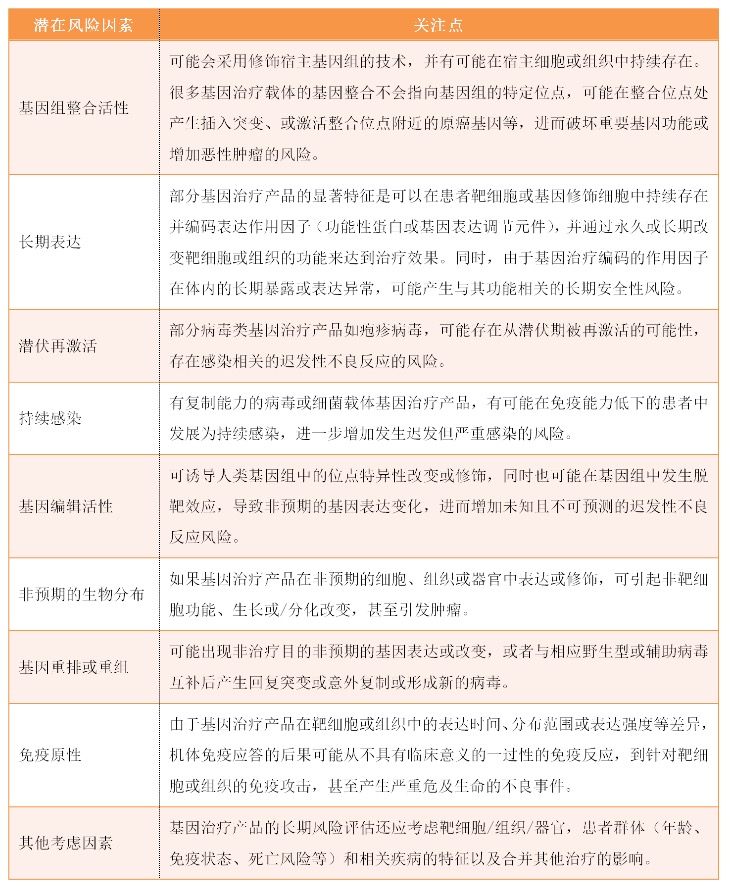
2、迟发性不良反应相关的潜在风险因素评估
在评估基因治疗产品的风险因素时,申请人应考虑基因治疗产品的特性,同时参考该产品的非临床和临床数据以及类似产品的已知数据,申请人应尽可能在非临床研究中获得用于评估迟发性不良反应风险的数据。包括但不限于以下几个要素:
3、临床研究人群相关的潜在风险因素评估
(1) 在设计长期随访临床研究的方案时,应考虑目标受试者人群及特征、整体健康情况以及接受治疗的患者的预期生存期等特征对迟发性不良反应的收集的影响。
(2) 当临床研究人群的某些特征(如预期寿命短、多重合并症、以及暴露于放疗或化疗等其他药物)可能干扰迟发性不良反应的观察分析时,会影响长期随访观察在评估和减轻受试者风险方面的效用;
(3) 而在病情较轻或较局限,合并症以及伴随治疗有限或较稳定的受试者中,通过长期随访观察收集到的评估数据可能更容易分析。
4、长期随访的设计实施
(1) 知情同意
1) 内容:需包含长期随访研究的目的、研究程序、持续时间、访视间隔以及研究者、伦理委员会或申办方的联系方式等。
2) 注意事项1:当非临床研究或临床试验中发现基因治疗产品的风险有所改变时,应及时更新知情同意书并告知受试者。
3) 注意事项2:知情同意书中还应对长期随访期间的人体组织样本采集和保存、基因检测等进行说明。
(2) 长期随访临床研究方案设计
1) 内容:应详细说明受试者的监测计划,包括访视时间表、采样计划、监测检查方法以及长期随访临床研究中的目标临床事件等。
2) 注意事项1:建议申办方提供一份简明科学的随访记录指导,供研究者及相关医务人员(包括研究者以外的医生和护士)记录所有观察结果和与研究相关的所有数据。如果在临床试验期间或上市后获得改变基因治疗产品风险的重要信息,应及时修订随访计划并予以实施。
(3) 长期随访实施
1) 在受试者接受基因治疗后的5年内(或根据具体产品的风险确定的长期随访期内),临床随访应记录受试者的简要病史,使用致癌或致突变药物和其他药物的情况以及有关的不良事件信息,新出现、复发或加重的疾病(例如恶性肿瘤、神经系统疾病、免疫原性或自身免疫类疾病、感染、甚至死亡等)及相关体格和实验室检查、受试者及其配偶的妊娠和生育情况等。同时,尽可能在合适的随访时间点采集相关样本,使用经过验证的、足够敏感的方法检测基因治疗产品在体内的持续存在情况并分析相关影响,直至数据表明不再有任何风险。如随访过程中出现疑似与基因治疗产品相关的不良事件,应及时根据临床、实验室、分子生物学、细胞遗传学、组织学或HLA分析获得的证据或深度测序数据等进行相关性的因果分析,必要时提高随访频率或增加随访内容。
2) 对于随访时间超过5年的基因治疗产品(或根据具体产品的风险确定的长期随访期内),完成前5年的随访后,可通过电话或书面调查问卷等方式,并尽可能采集相关样本,保持每年至少随访受试者一次直至随访期结束。如前期随访提示产品在体内持续存在,建议观察至数据表明不再存在任何风险。
参考文献
[1] http://www.cde.org.cn


