今天的新药发现,已经离不开计算学科的支撑,与计算相关的各种技术也因新药研发,而备受行业的重视。机器学习,作为AI的一个重要分支,凭借其辅助发现潜力化合物、预测相关参数、节约试验成本、压缩开发周期等优势,得到了研发及投行的极大关注。本稿件即对机器学习的历史及其于医药领域的应用进行概述,以期与同行进行共同学习。

未来:精准医学&药物发现
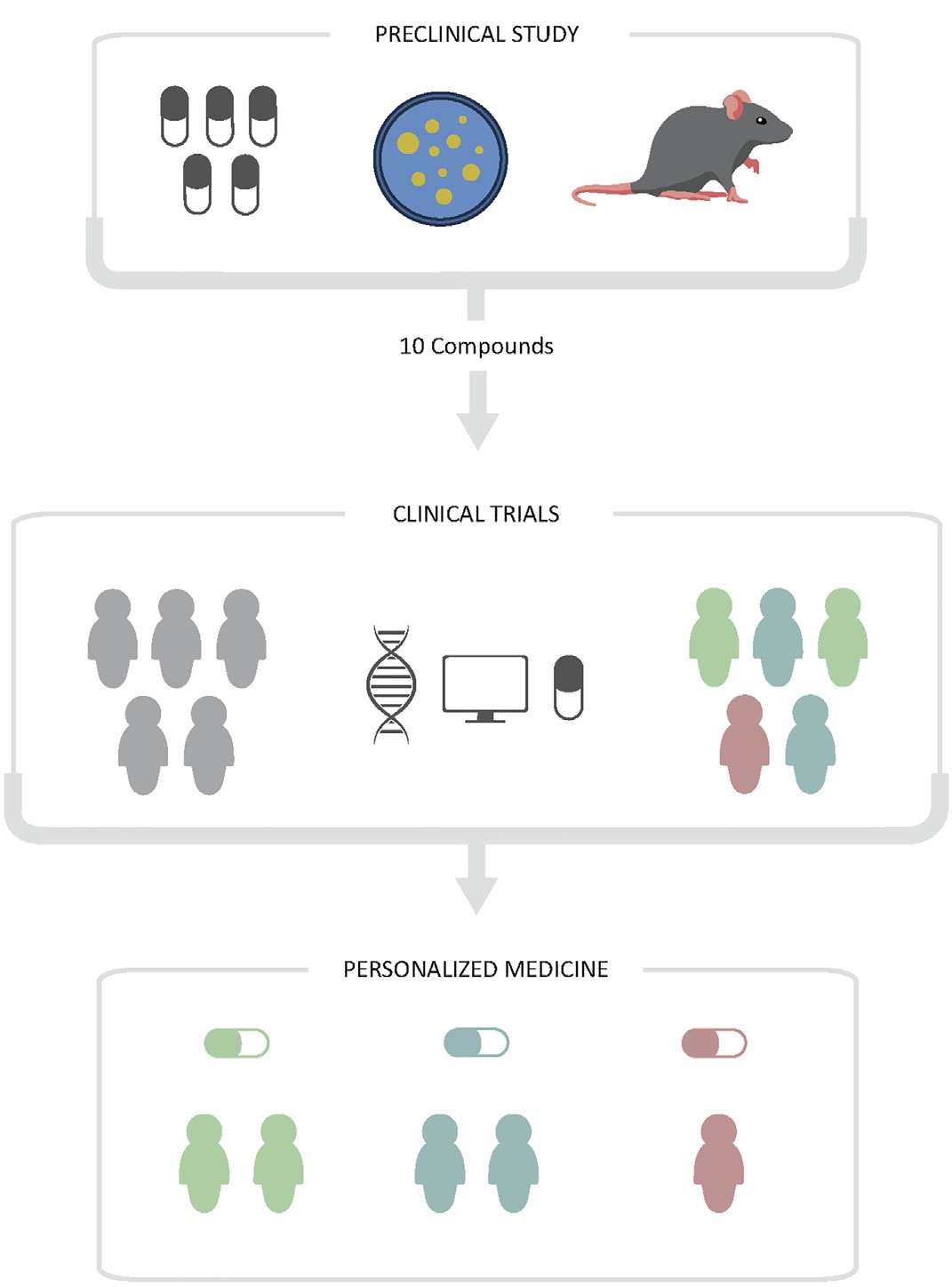
近年来,精准医学的概念越来越被提及,其强调基于个体差异(包括基因&环境&生活方式等)来进行疾病的预防和治疗,从而降低“一刀切”的治疗方式。出于这个原因,近年来产生了大量生物医学数据,其来源非常多样化:从小型的实验室到大型的多中心研究;这些数据主要称为组学数据(基因组学、蛋白质组学、代谢组学、药物基因组学等),是科学界取之不尽的信息来源,可用于对患者进行分类,获得特定诊断,以及开发新的治疗方法。
过去十年中,计算能力的快速提升,已逐渐形成了与传统药物发现过程中高通量筛选的竞争。机器学习(ML),作为人工智能的一个分支,已有多重方法应用于药物发现过程中,从而预测新化学实体的分子特征、生物活性、相互作用和不良反应等。这些算法,正在改变着新药发现的传统模式。
图1.1 精准医学背景下-新药发现过程(见参考文献)
ML在Drug-Discovery领域的发展
ML在Drug-Discovery领域的发展
1964年,Hansch方程的提出,理化描述符(如疏水性参数、电子参数和空间参数)的线性回归模型,开始用于描述二维结构-活性关系,QSAR的概念逐渐深化发展。
1998年,类药性概念的提出,研究者开始建立可以高效预测分子是否具备药物潜质的模型,并从1D/2D描述符中慢慢深入。但总的来说,2000年以前,ML于药物发现领域的应用,并不多,主要原因是数据的可用性问题。
2004年,PubChem和ZINC数据库的开发,为ML于药物发现的发展奠定了基础;并在2006年和2008年开发了DrugBank和ChEMBL,从而大大满足了上述的数据可用性问题。
2016年,Molecular Graph Convolutions正式发布,相关研究人员的成果也于2020年在Cell杂志刊登,进一步展示了机器学习在该领域的潜力,并发现了一种具抗菌活性的分子halicin,并在实验室中得到了验证。

图2.1 药物发现领域-机器学习主要事件时间表(见参考文献)
ML操作流程
药物发现领域的ML方法,涵盖以下步骤:1)数据收集;2)数学描述符的生成;3)搜索变量的最佳子集;4)模型训练;5)模型验证。

图3.1 药物发现-机器学习方法(见参考文献)
如上所述,首先是收集数据,数据除了有助于活性、选择性、代谢、毒性、理化性质外,甚至还需要易于生产制备等属性;小分子和肽类药物,可以使用SMILES和FASTA格式表示结构的序列;数据库如DrugBank、PubChem、ChEMBL、ZINC等,具有大量的数据储备信息。随着数学描述符(PCA、t-SNE、FS、Autoencoder相关技术)的生成,可获得一系列的数据,ML模型即可以处理这些数据。数据可分为两个子集,高比例数据用于模型训练,低比例数据用于测试,这一过程可获得变量相关的最佳子集。在模型训练后,可依此而完成后续的验证,如果验证结果具有统计意义,可以说,即创造了一种新的药物预测模型。PS:最好的模型是以最低的总成本实现最高的性能价值。
输入数据-极其重要
模型的训练,至关重要的一个环节就是具代表性描述特性的分子描述符的输入,进一步相关QSAR、分子描述符、计算信息指纹、基于图的机器算法,等等。
➢ QSAR
QSAR,是通过结构与活性的关系以数值的形式进行关联;即通过整合计算和统计,对生物活性进行理论预测,从而可以对未来可能的新药进行理论设计,理论上节省了研发成本。要进行QSAR研究,需要3类信息:1)具有共同作用机制的不同化合物的分子结构;2)每个配体的生物活性数据;3)理化性质。
➢ 分子描述符
MD,即定量描述相应理化性质的分子的数字表示;依此,研究者可根据与计算描述符数值的相似性来找到具有相似物理化学性质的分子。分子描述符可分为两大类:1)实验测量值,如logP、偶极矩、极化率等;2)理论值,如结构、拓扑、几何、电子、理化等等。理论分子描述符又可以根据其维度建立0D/1D/2D/3D/4D/5D/6D描述符,其中3D/4D的研究最为深入。
➢ 计算信息指纹
FP,是一种特殊形式的分子描述符,通过具有固定长度的位向量快速有效表示分子结构,以表明内部子结构或官能团的存在或不存在。不过,源自化学结构的指纹忽略了生物特征,从而在分子结构和生物活性之间关联度度降低,以至于前者的微小变化都会产生生物活性的实质性差异。FP在计算工作中,常常关联MACCS、Pubchem、CDK等。
➢ 基于图的机器算法
化合物结构式在图方面的表示,主要为分子网络,网络中的每个原子都表示为网络中的一个节点,使用的算法主要为人工神经元网络。早在2009年,即有研究者提出了图神经网络模型;2016年,斯坦福大学和谷歌公司的研究人员开发了分子卷积图,而正是由于将卷积算法应用于图形,药物发现中的计算研究向前迈进了一步。
ML&生物学问题
现代生物学的复杂性,使计算成为支撑生物学实验必不可少的工具,因为它们允许以高精度编码理论模型来处理大量信息,从而促进和加速新药的开发。无论是从hit-to-lead,还是一定程度的ADMET,计算都能给出一定的预测。通过抽取2016-2020年的文章样本,统计相关生物学问题如下。

图5.1 2016-2020年样本文章解决的生物学问题(见参考文献)
如上所述,比例最高的为“药物-靶标相互作用”。靶标研究,位于疾病和药物发现的最前端,这个“开头”的重要性,自不必说。化合物-蛋白相互作用,已成为新药发现的先决条件,如PDB数据库的使用,通过积累大量的受体-配体结晶,为相互作用提供了大量的数据,是药物计算研究人员必不可少的数据来源,同时,相应的也诞生了许多进行测算的软件,如MPLs-Pred。
ML未来发展趋势
贝叶斯、支持向量机、决策树、人工神经网络的深入研究,无疑会为机器学习的精准度大大助力;而基于结构的药物设计,将更加离不开机器学习,从而达到快速、高效、低成本的行业要求。然而,机器学习的优点已有大量研究进行展示,但不得不说的是,真正凭借机器学习、人工智能为核心技术,而开发出的上市药物,还没有。故,基于机器学习的药物发现,也一直受到行业的质疑。但技术上的重大突破,往往伴随着前期的极度质疑,而一旦实现质的飞跃,也必将受到更大的投资回报。机器学习,人工智能,正在发力,未来可期!
参考文献:
1.review on machine learning approaches and trends in drug discovery. doi.org/10.1016/j.csbj.2021.08.011
2.AI-based language models powering drug discovery and development. doi.org/10.1016/j.drudis.2021.06.009
3.Integration of AI and traditional medicine in drug discovery. doi.org/10.1016/j.drudis.2021.01.008


